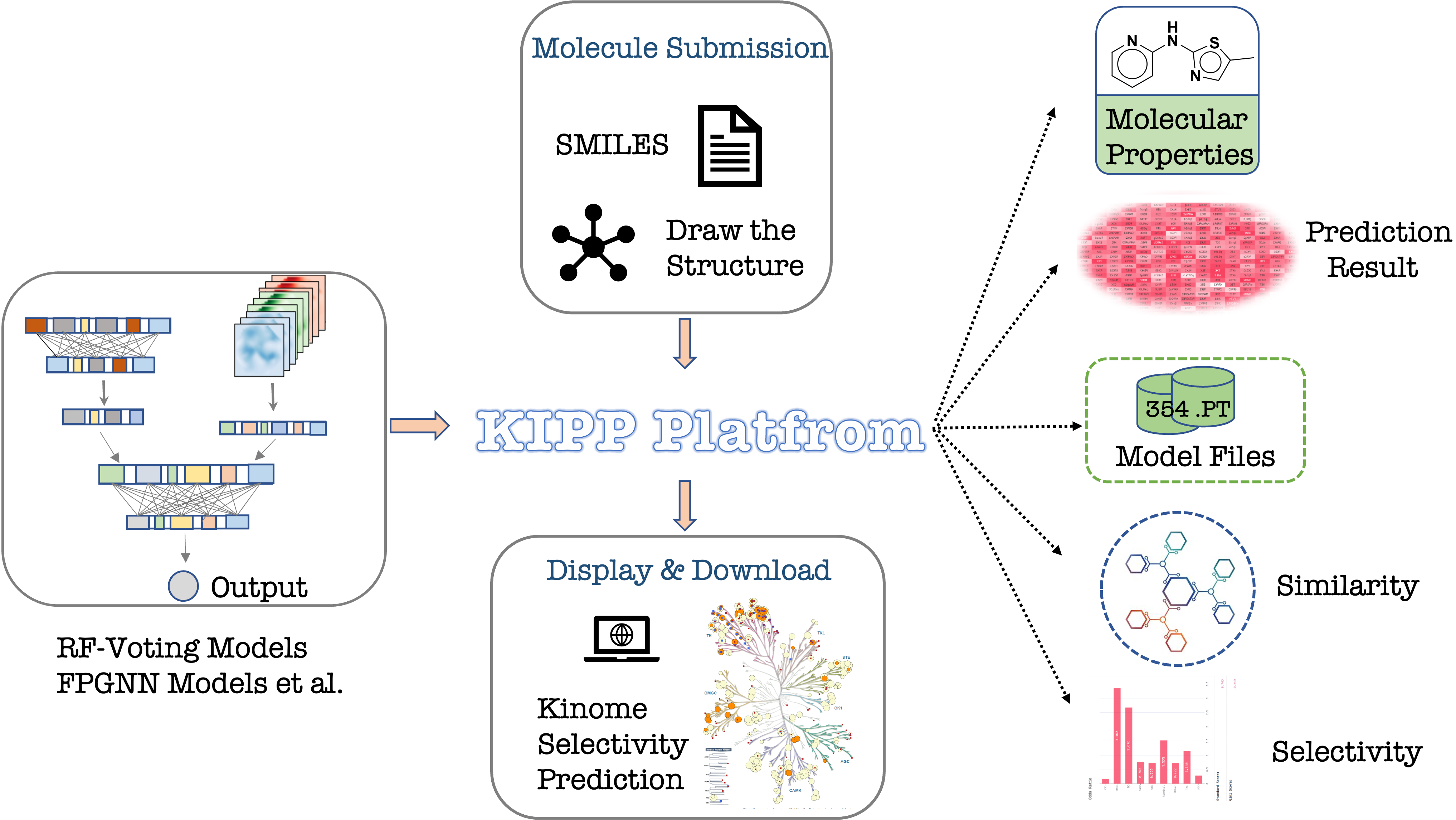
What is KIPP ?
The selectivity of kinase inhibitor for the kinase profile is critical for the discovery and development of drugs targeting kinases. Currently, machine learning algorithms are widely used in kinase inhibitor profiling prediction due to their fast, accurate and low consumption characteristics.
By comparing different molecular features (molecular descriptors, fingerprints and graphs) as well as the popular conventional machine learning or advanced deep learning algorithms, an online platform KIPP was developed based on the overall optimal Voting-RF-RDKitDes+FP2+AtomPairs models (default) as well as a collection of the best models based on each kinase (optional) for the large-scale kinome-wide virtual profiling for small molecules.
Through a comprehensive analysis of the kinome-wide inhibitory activity for a given small molecule, KIPP can depict the overall selectivity and the selectivity towards a subfamily of kinases based on the predicted kinase profile, which enables users to create a comprehensive network of excimer interactions for the design of new chemical modulators targeting a specific kinase.
Molecular representations adopted in this research are as follows:
(1) Morgan fingerprints (ECFP-like, 1024-bits) is transformed from the standard Morgan
algorithm, and the radius is set to 2, which is equivalent to the ECFP4 fingerprint.
(2) MACCS keys (Molecular ACCess System, 166-bits) is derived from the chemical
structure database developed by MDL. The original length is 166 bits. It encodes specific
structural patterns through SMARTS and belongs to dictionary-based fingerprints.
(3) AtomParis fingerprints (1024-bits) is a simple and ancient fingerprint, whose atom
types are all composed of elements, the number of adjacent atoms of heavy atoms and the
number of 𝜋 electrons.
(4) FP2 fingerprints is a path-based 1024-bit 2D fingerprint that resolves molecular
structure, identifying linear fragments of 1-7 atoms in length to index small molecule
fragments.
(5) PharmacoPFP is a 2D Pharmacophore Fingerprints (38-bits). It usually encodes the
structural features of molecules in a similar way to substructure-based fingerprints, but
can also consider the distances between these features, and classify them according to the
distance range to generate bit strings. Pharmacophore fingerprints can be used to analyze
similarities between molecules or between molecular libraries.
(6) Descriptor-based methods use quantitative molecular descriptors (including molecular
physicochemical properties and molecular composition) as input features to build QSAR
models. In this study, 208 RDKit descriptors including molecular weight, lipid-water
partition coefficient, Topological Polar Surface Area (TPSA), the number of donor hydrogens,
the number of acceptor hydrogens, the number of benzene rings, and the number of functional
groups were used to construct descriptor-based models.
The dataset of KIPP can be downloaded here: KinaseNet.rar, KIPPRawData.rar
The SHapley Additive exPlanation (SHAP) interpretability of the dataset: Kinase_SHAP_Interpretation.rar
Validation of model application in selective prediction: Table 1 Table 2 Table 3